Crop epigenetics and epigenomics
Epigenetic inheritance
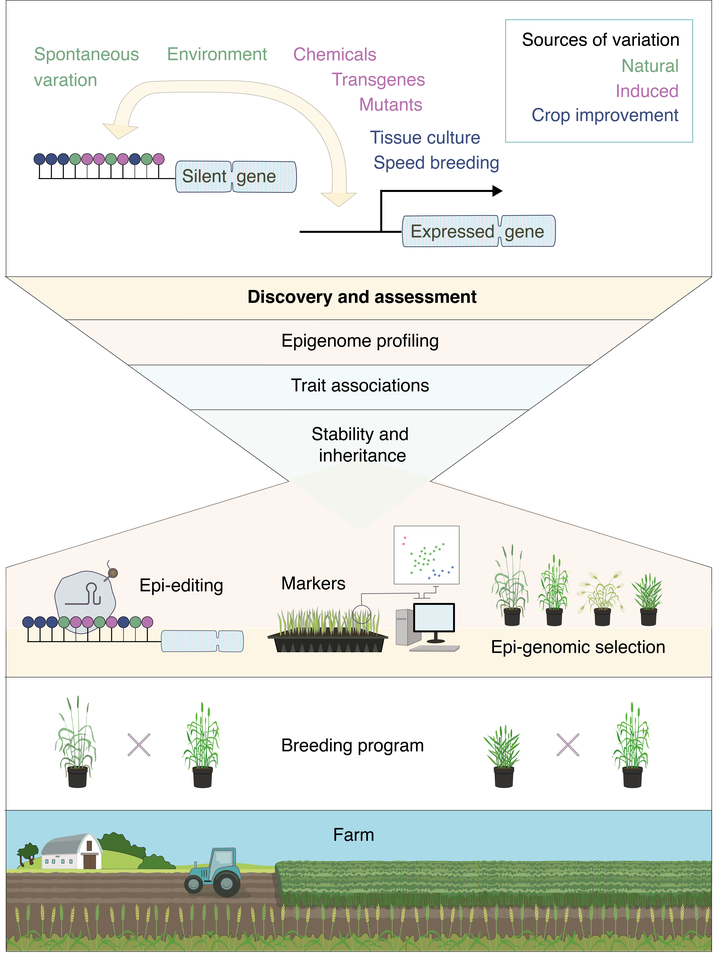
Technological advances in sequencing throughput and epigenome editing are enabling us to now readdress fundamental unanswered questions concerning the inheritance, stability, variation and functional relevance of epialleles in large crop genomes. Such investigations are having a profound impact on our understanding and the utility of epigenetic information for agriculture.
A major focus of our research is characterizing the inheritance patterns of DNA methylation and other epialleles, which contribute to the unexpected and non-mendelian inheritance of traits. We recently studied in detail maize plants that were passed through tissue culture (eg for transformation) and found accumulation of targeted, heritable changes in DNA methylation at unstable loci (Han et al., 2018). We are also continuing to study the inheritance of DNA methylation over generations in maize populations.
In contrast to maize, clonal horticultural species, like apple, present a unique opportunity to study and potentially exploit epigenomic regulation and inheritance within a single genetic lineage. We are tracking the inheritance and variation of epialleles in clonal apple lineages, which could relate to spontaneous variation and trait stability.
Epigenome engineering in monocot crops
Natural variation is a powerful resource for investigating the role of DNA methylation. However, using epigenome engineering tools including CRISPR, we are working to generate germplasm with far greater levels of epigenetic variation.
Currently, in crops species with large genomes, germplasm with even moderate reductions in DNA methylation are not available, severely limiting our ability to study the role of epialleles. We are also implementing tools for locus-specific epigenome editing in monocots. Ultimately, reengineering the levels of methylation will allow us to answer fundamental questions about the function of methylation throughout crop genomes. Future applications also include using epigenome engineering and synthetic biology to modulate epialleles for crop improvement.
Cryptic epigenetic variation
In large crop genomes there are many silent loci, including protein coding genes, which are associated with DNA methylation (Eichten et al., 2013). These loci may represent cryptic variation. The extent to which these loci are expressed in a subset of members of a species is relatively unexplored in crop genomics due to the historical costs of sequencing technologies.
We are identifying cryptic variation in barley using comparative (epi)genomics in combination with the introduction of epigenome engineering tools. In the longer term we will use these varieties to develop Epigenetic Recombinant Inbred Lines (epiRILs) to uncover cryptic epialleles associated with trait variation through activating silent parts of the genome.
Publications
Beyond the gene: epigenetic and cis-regulatory targets offer new breeding potential for the future