Epigenome guided crop improvement - distilling the Functional Genome
Using epigenomic signatures to discover ‘functional’ and cis-regulatory elements in large crop genomes
Many crops have incredibly large genomes, ranging from the modest 730 megabases (Mb) sorghum genome to the massive 17,000 Mb hexaploid wheat genome. Yet, recently developed technologies have enabled even the larger genomes to be completely sequenced and have provided unprecedented insights into the number, form and function of their genes and the landscape of their chromosomes.
The identification of sequences of protein-coding genes and non-coding RNAs, with their associated promoter, terminator and intron sequences, has become relatively straightforward and accurate. However, it is becoming increasingly clear that there are other regions within the genome that play an enormous role in regulating and coordinating gene expression. These regions are not easily identified bioinformatically, as they are often located a considerable distance from their target genes and do not have readily-distinguishable sequences.
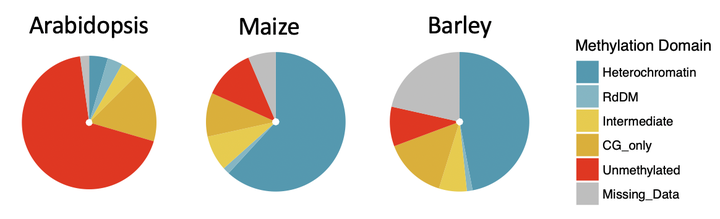
Together with collaborators (Waterhouse lab, QUT, Australia; Schmitz Lab, UGA, USA; Springer lab, UMN, USA ), we are developing a novel methodology to efficiently and affordably identify gene control regions, known as cis-regulatory elements, in large genome crops. Our preliminary analysis, together with prior studies in plants, suggests that cis-elements are predictably marked by a distinct lack of DNA methylation; we term these “unmethylated regions” (UMRs) (Crisp et al., 2019). We are testing the hypothesis that DNA methylation profiles can be used to efficiently identify cis-elements in plant genomes. Key elements can then be targeted for crop improvement using CRISPR and other genome editing approaches.
In this project, we are leveraging the resources of N. benthamiana (tobacco), which is peerless in its amenability to functional validation work (eg CRISPR) and transient expression analysis; in combination with methylC-seq, ATAC-seq, STARR-seq and RNA-seq.
This project initially focused on N. benthamiana and maize, and is now expanding to realise the tremendous opportunities many other crop species including in sorghum, barley and wheat.
Publications
Beyond the gene: epigenetic and cis-regulatory targets offer new breeding potential for the future